The major goals of our research are the development and application of new synthetic methods in organic chemistry. The focus is on catalytic methods, which enable previously unknown transformations employing both photochemical and conventional techniques. Our research is curiosity-driven and does not strive for immediate industrial applications. However, entrepreneurial opportunities are also considered where applicable.
Natural Product Synthesis
The selection of natural product targets in our group is based on aspects of structural uniqueness, the application of suitable new methodology and their biological activity. In the course of our work, we have been able to elucidate the constitution/composition and configuration of several natural products by accomplishing their first total syntheses. Examples include the syntheses of wailupemycin B, punctaporonin C, lactiflorin, and pinolinone.
New synthetic methods, which were developed in our group, have been successfully applied to the synthesis of natural products. The synthesis of meloscine, for example, was the first synthesis to employ an enantioselective photochemical key step in natural product synthesis. Additionally, a C-H activation protocol, which we developed for addressing the C2 position of the indole core, was applied to the synthesis of the aspidosperma alkaloids aspidospermidine and goniomitine. Our interest in diastereoselective reactions of carbenium ions led to a concise synthesis of podophyllotoxin.
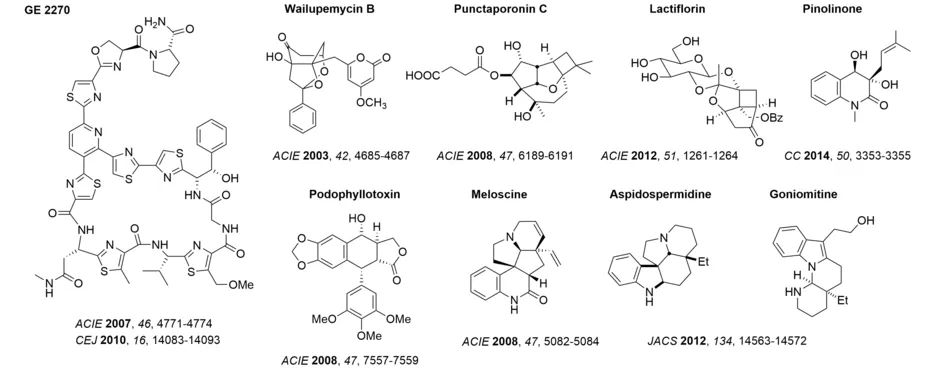
Biological activity is a further motivation to approach a synthetic target. In this regard, our research has been devoted to the synthesis of anticancer and anti-infective compounds. To highlight an example, the syntheses of the GE-factors and the amythiamicins are stated above, which address the bacterial elongation factor EF-Tu. Several of these studies have been and are performed in collaborations. We successfully identified the mode of action of some modified EF-Tu inhibitors (ChemMedChem 2013, 8, 1954-1962) and we contributed to the elucidation of the vioprolide biosynthesis pathway (Angew. Chem. Int. Ed. 2018, 57, 8754-8759). More recently, we completed the first total synthesis of a vioprolide, vioprolide D, the sterically demanding (E)-dehydrobutyrine fragment of which was established in the final stages of the synthesis by an isomerization of the (Z)-compound. Studies towards EF-Tu culminated in the successful total synthesis of a further, structurally unique inhibitor of this protein, pulvomycin D, a polyketide natural product. In this instance, a stereoselective aldol reaction facilitated the convergent assembly of the molecular skeleton (Synthesis 2021, 54, 4246-4262) and the cyclization of the macrocyclic lactone was accomplished by an intramolecular Heck reaction. The synthesis of the enigmatic polyketide enterocin established a link to previous studies on the biogenetically related wailupemycin B. Enantiomerically pure material was secured by choice of a selective, biomimetic synthesis route, which in turn allowed for an assignment of its absolute configuration (Org. Lett. 2022, 24, 6903-6907).
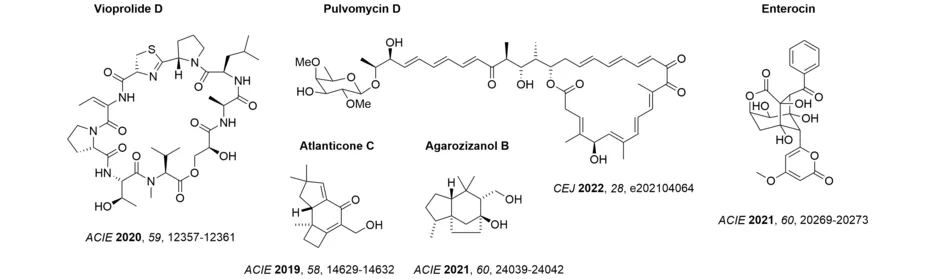
The discovery of an unprecedented cascade reaction in which up to three photochemical steps were combined, enabled an access to sesquiterpenes with a protoilludane or prezizaene skeleton. The utility of the method was demonstrated by the first total syntheses of the protoilludane atlanticone C and of the prezizaene agarozizanol B.
In addition to our efforts towards the synthesis of natural products, there is a focus on the preparation of new scaffolds for medicinal chemistry (e.g. Angew. Chem. Int. Ed. 2012, 51, 10169-10172; Nat. Commun. 2020, 11, 5621) and on the development of new probes for biological studies (e.g. J. Am. Chem. Soc. 2018, 140, 2718-2721). These projects are mostly performed in collaboration with other academic and industrial institutions.
Catalytic Methods
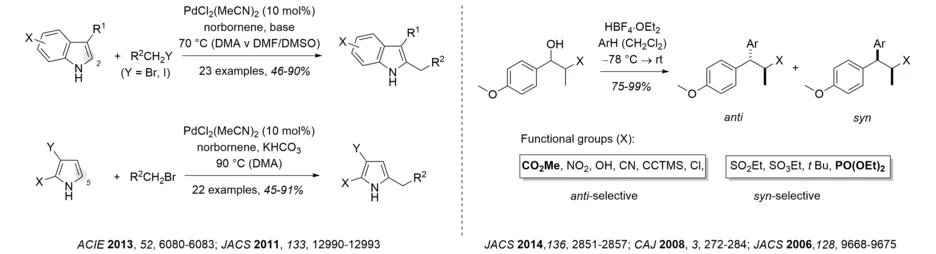
Our interest in the synthesis of heterocycles has led to the development of regioselective cross-coupling reactions, which in turn have found widespread applications (Review: Tetrahedron 2005, 61, 2245-2267). In recent years, interest has shifted towards the direct C-C bond formation on heterocyclic cores by C-H activation reactions. Examples include the alkylation of indoles and pyrroles and the arylation of thiophenes (e.g. Chem. Eur. J. 2015, 21, 18407-18416).
Our group was the first to comprehensively and systematically establish and develop the facial diastereoselectivity in the intermolecular reaction of free carbenium ions. It was shown that the reaction of benzylic cations proceeds with high diastereoselectivity and that the outcome depends on the steric size of the respective substituents at the stereogenic center in α-position of the cation. Catalytic versions of these transformations were developed employing FeCl3, AuCl3 or Bi(OTf)3 as catalysts. The study has been further extended to allylic and propargylic cations (e.g. Angew. Chem. Int. Ed. 2008, 47, 10106-10109).
Initial work in the area of hydrogen-bonded catalysis was dedicated towards finding enantioselective catalysts for photochemical reactions (see below). Our interest in this topic has further expanded towards enantio- and regioselective transition metal catalysis (Review: J. Org. Chem. 2019, 84, 8815-8836). Therefore, we designed ligands, which bear a site for substrate binding via hydrogen bonds and which also enable the attachment of catalytically active transition metals, such as Mn, Ru or Rh. Proof of principle studies were performed using a Ru-based oxidation catalyst and quinolone-based olefins for selective epoxidation reactions. It was demonstrated with high confidence that hydrogen bonding is responsible for both regio- and enantioselectivity. This Rh catalysis allowed us to achieve enantioselective amination and aziridination. A more recent development relates to Mn-based catalysts which allow for site- and highly enantioselective oxygenation reactions (Angew. Chem. Int. Ed. 2018, 57, 2953-2957; Chem. Sci. 2020, 11, 2121-2129).
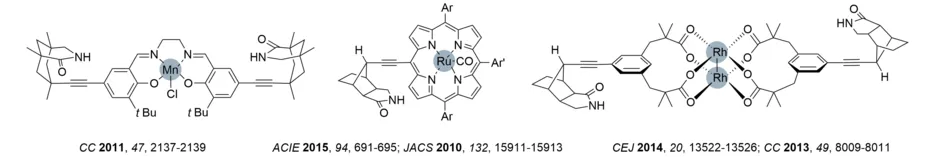
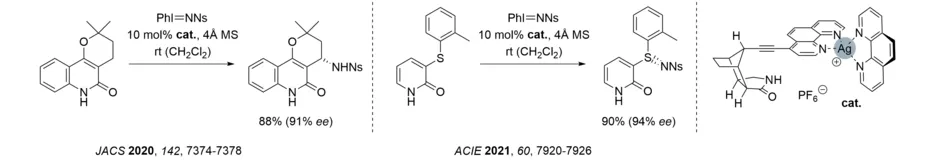
The covalent linkage of a phenanthroline to the established octahydro-1H-4,7-methanoisoindol-1-one skeleton enabled the in situ preparation of a heteroleptic silver complex. The protocol allowed for the first enantioselective silver-catalyzed amination and sulfimidation reaction (Ns = p-nitrophenylsulfonyl). The substrates were either quinolones or 2-pyridones.
Given that our goal is to develop synthetic methods for practical applications, we strive to show an immediate relevance of our methods for total synthesis. Recent examples include a convenient C-H amination reaction (Adv. Synth. Catal. 2016, 358, 2083-2087) or a new pyrrole synthesis (J. Org. Chem. 2016, 81, 6149-6156), which were both applied to the synthesis of heterocyclic alkaloids, as well as a Rh-catalyzed arene hydrogenation for the diastereoselective preparation of 2,5-diketopiperazines (ACS Catal. 2022, 12, 3628-3633).
Photochemistry
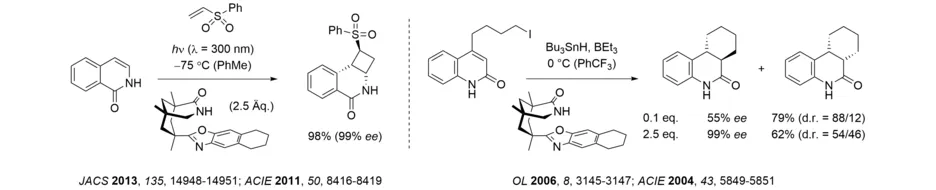
Ever since it has been established, our group has tackled unsolved challenges in photochemistry. Since the year 2000, a particular emphasis has been on the preparation of enantiopure compounds by photochemical methods, and we have made fundamental contributions to the field of enantioselective photocatalysis. Based on the 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonane-2-one scaffold, which is readily derived from Kemp’s triacid, we have developed a chiral template for photochemical and radical reactions, which has proven its versatility over the last 20 years. Interestingly, the template showed some enantioselective catalytic activity in radical reactions. In recent years, it has been successfully employed for enantioselective intra- and intermolecular [2+2] photocycloaddition reactions of isoquinolones.
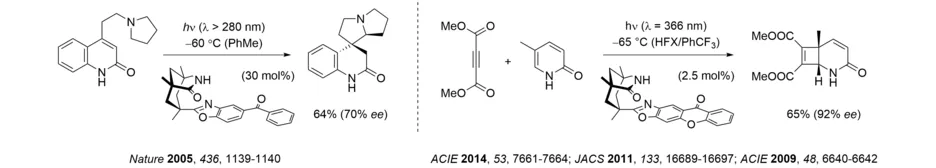
Upon modification of the oxazole backbone, it was possible to develop the template into enantioselective catalysts, which work either by electron transfer or by energy transfer (Review: Chem. Rev. 2022, 122, 1626-1653). A ketone served as the catalyst in the enantioselective photoredox cyclization of an aminoethyl quinolone. A xanthone was employed for the enantioselective sensitization of [2+2] photocycloaddition reactions with catalyst loadings as low as 2.5 mol% (HFX = 1,3-hexafluoroxylene).
A recent development in this area relates to thioxanthone sensitization which can be performed very efficiently with artificial visible light sources or with sunlight. It was found that a chiral thioxanthone enables inter- and intramolecular [2+2] photocycloaddition reactions of quinolones and, most remarkably, the deracemization of allenes. The latter reaction allows for a thermally impossible process, i.e. the formation of single enantiomer from a racemic mixture, and holds great promise for further applications. Its mode of action relies on a complex interplay of several parameters including catalyst association, sensitization efficiency and intermediate decay.
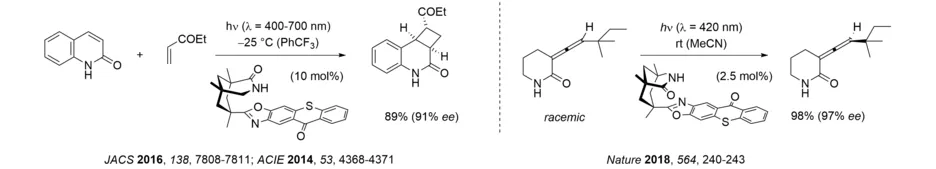
After we first reported an efficient photochemical deracemization in 2018, interest in this topic has grown rapidly. In our group, research is focused on the development of reactions, in which a chiral catalyst differentiates between the two enantiomers of the substrate in the first photochemical step. Since – in an ideal scenario – only one enantiomer is processed and a racemization occurs, the other enantiomer is enriched without loss of material. In the case of axially chiral allenes and alkenes, triplet energy transfer from a thioxanthone facilitates the formation of an achiral intermediate which forms either one of both enantiomers when returning to the ground state hypersurface. Based on this concept, it was possible to deracemize both acyclic allene amides as well as axially chiral alkenes.
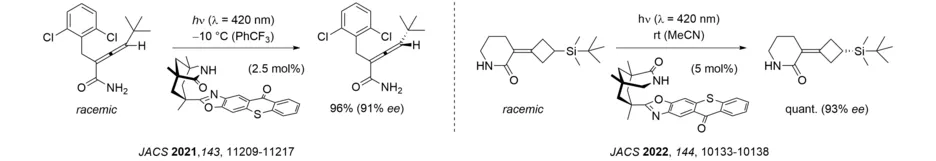
The photochemical deracemization at stereogenic carbon centers requires the reversible fission/cleavage of single bonds. We could show that cyclopropanes can undergo such a bond cleavage upon triplet energy transfer (Angew. Chem. Int. Ed. 2019, 58, 3538-3541) and we could spectroscopically detect the 1,3-diradical responsible for the deracemization of spirocyclopropyl oxindoles. Hydantoins can be obtained in high yield and enantioselectivity by a photochemical deracemization that involves a reversible hydrogen atom transfer to a chiral benzophenone catalyst.
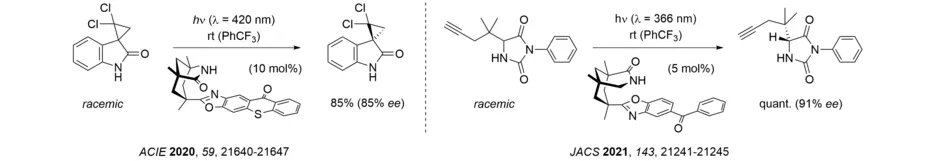
In 2010, Lewis acid catalysis was identified as a suitable method to achieve enantioselective photochemical reactions, as demonstrated by the intramolecular [2+2] photocycloaddition of coumarins. In 2013, it was shown that enantioselective Lewis acid catalysis is not restricted to the coumarin [2+2] photocycloaddition but can – unexpectedly – also be applied to enone [2+2] photocycloaddition reactions. The high enantioselectivity is unexpected because enones – unlike coumarins - undergo uncatalyzed [2+2] photocycloaddition reactions at λ = 366 nm. The mode of action in the enone case is clearly different from the coumarin case. Enantioselective Lewis acid catalysis of enone [2+2] photocycloaddition seems to be relatively general and was applied not only to dihydropyridones but also to 2-cycloalkenone substrates (Review: Acc. Chem. Res. 2020, 53, 1933-1943).

It has been shown that the disadvantage of high catalyst loading can be overcome if a substrate that is unbound to the Lewis acid and its Lewis acid complex show no overlap in the long-wavelength region of the UV/Vis spectrum. If this requirement is fulfilled there is no racemic background reaction and the enantioselectivity remains high even at low catalyst loading. The visible light-mediated, intermolecular ortho photocycloaddition to phenanthrene-9-carboxaldehyde serves as an example. A similarly low catalyst loading can be utilized if the relevant reaction occurs at the singlet hypersurface and a background reaction of the enone via the respective triplet can be excluded. Such a process was observed for the oxadi-π-methane rearrangement of 2,4-cyclohexadienones and was used for the enantioselective synthesis of chiral cyclopropanes.
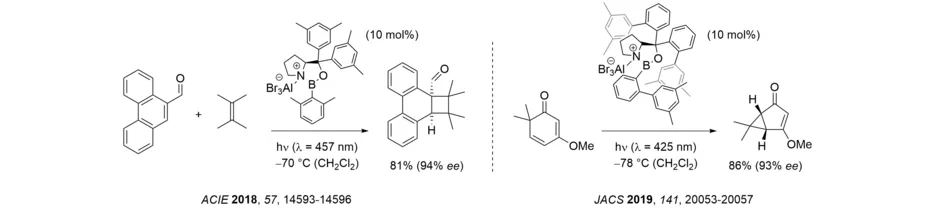
The latter examples illustrate how a chromophore can be activated towards a selective reaction at long wavelength and studies along these lines continue in our laboratory (Review: Angew. Chem. Int. Ed. 2018, 57, 14338-14349). After the discovery of a Brønsted acid-catalyzed [2+2] photocycloaddition (Angew. Chem. Int. Ed. 2017, 56, 4337-4341) it was shown that the generation of iminium ions facilitates a triplet-sensitized process and that the intermolecular [2+2] photocycloaddition of iminium ions can be performed enantioselectively. The energy transfer from the ruthenium complex (bpy = 2,2‘-bipyridine) to the iminium ion was detected by time-resolved spectroscopy and an electron transfer was ruled out. Generally, it has been shown that iminium ions display a lower triplet energy transfer than the carbonyl compounds they are derived from, which in turn enables a facile excitation by energy transfer. A chiral phosphoric acid displaying two thioxanthones units was successfully employed in the catalytic intermolecular [2+2] photocycloaddition of N,O-acetals which also proceeds via iminium ion intermediates.
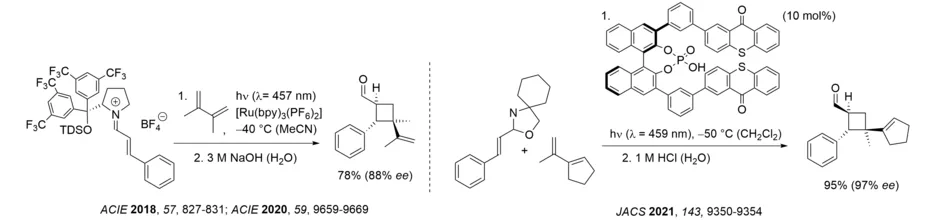
One of the most remarkable features of photochemical reactions relates to the fact that molecular scaffolds become available that are very difficult or even impossible to obtain thermally. In this context, research in our group is concerned with photochemically induced reaction cascades in which several photochemical steps are performed in a row or in which a photochemical step triggers subsequent rearrangement reactions. Below three examples are shown (from left to right): a three-photon reaction cascade starting from a 1-indanone, a formal intramolecular carboformylation of an olefin, and the Lewis acid-catalyzed formation of benzoisochromanes from 1-naphthaldehydes. In all three instances, it is an intramolecular ortho photocycloaddition that initiates the observed reaction cascade. The reaction of the iminium ions can also be performed with visible light and a suitable triplet sensitizer. The reaction cascade of 1-naphthaldehydes occurs in much higher yields and with excellent enantioselectivity if performed with a chiral oxazaborolidine as Lewis acid catalyst.
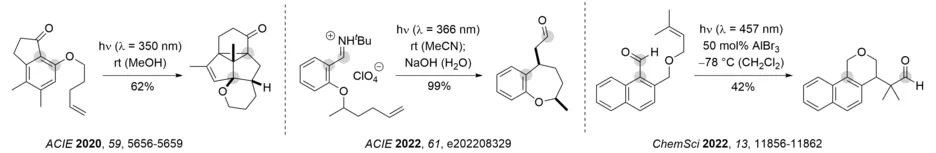
In photochemistry, we combine synthetic experiments with photophysical studies which are partially performed in our laboratory and partially in collaboration with other groups. Beyond the above-mentioned examples, our studies on the activation of fundamental chromophores with a Lewis acid deserve to be explicitly mentioned. In a combined effort by synthesis, spectroscopy, and theory, the photochemical reaction network of the 2-cyclohexenone-BF3 (Angew. Chem. Int. Ed. 2021, 60, 10155-10163) and of the benzaldehyde-BCl3 complex was elucidated (J. Am. Chem. Soc. 2022, 144, 18927-18937).
Funding
Our research continues to be supported by TU München and the state of Bavaria. Third party funding is provided mainly by the Transregio 325 (https://crc325.de) and the Gottfried Wilhelm Leibniz Award. Both programs are administered and funded by the Deutsche Forschungsgemeinschaft (DFG). Until December 2020, our research was supported by the European Research Council (ERC) in the framework of the Horizon 2020 research and innovation programme (grant agreement No 665951 – ELICOS). Additional institutions which support us or have supported us over the years include the DFG (individual research grants, GRK 1626), the Fonds der Chemischen Industrie, the Alexander von Humboldt Foundation, the German Academic Exchange Service (DAAD), the Elitenetzwerk Bayern, the Federal Ministry of Education and Research (BMBF) and the Studienstiftung des Deutschen Volkes. We continuously collaborate and have collaborated with many companies on joint research projects, inter alia with (in alphabetical order): AstraZeneca, BASF, Bayer, Bicoll, Evonik, Medigene, Novartis, Roche, Sanofi, and Wacker.