Forschung
Unsere Forschung ist hauptsächlich auf die Entwicklung und Anwendung neuer synthetischer Methoden innerhalb der Organischen Chemie ausgerichtet. Der Fokus liegt dabei auf katalytischen thermischen sowie photochemischen Verfahren, um neuartige Transformationen zu ermöglichen. Unsere Forschung wird durch akademische Fragestellungen getrieben und zielt nicht unmittelbar auf industrielle Anwendungen. Jedoch werden unternehmerische Möglichkeiten genutzt, wo inhaltlich sinnvoll.
Totalsynthese
Die Auswahl von Naturstoffen als Zielverbindungen basiert hauptsächlich auf Aspekten der strukturellen Einzigartigkeit, der Anwendbarkeit neuer Methodik sowie der biologischen Aktivität. Durch Erstsynthesen konnten wir bisher die Konstitution und Konfiguration mehrerer Naturstoffe bestätigen, unter anderem von Wailupemycin B, Punctaporonin C, Lactiflorin und Pinolinon. In unserer Gruppe entwickelte neuartige Synthesemethoden konnten erfolgreich in Totalsynthesen von Naturstoffen angewandt werden. So wurde in der Synthese von Meloscin zum ersten Mal ein enantioselektiver photochemischer Schlüsselschritt verwirklicht. Mit Hilfe eines von uns entwickelten Protokolls zur C-H-Aktivierung konnte die C2-Position des Indol-Kerns adressiert werden, um so zu den Aspidosperma-Alkaloiden Aspidospermidin und Goniomitin zu gelangen. Unser Interesse an diastereoselektiven Reaktionen von Carbeniumionen führte zu einer effizienten Synthese von Podophyllotoxin.
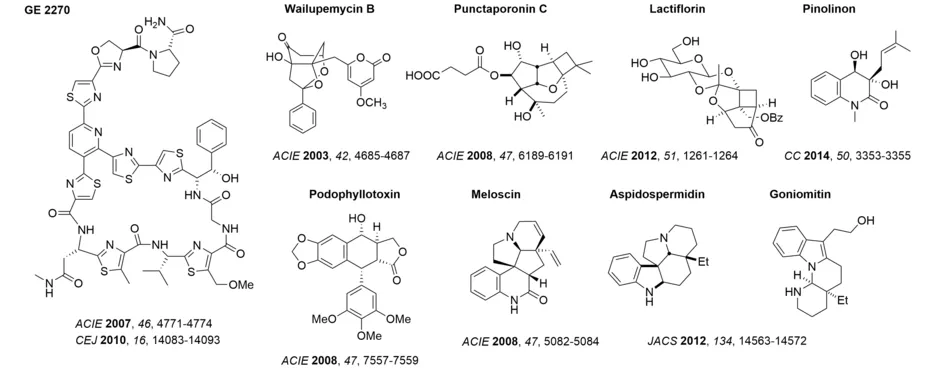
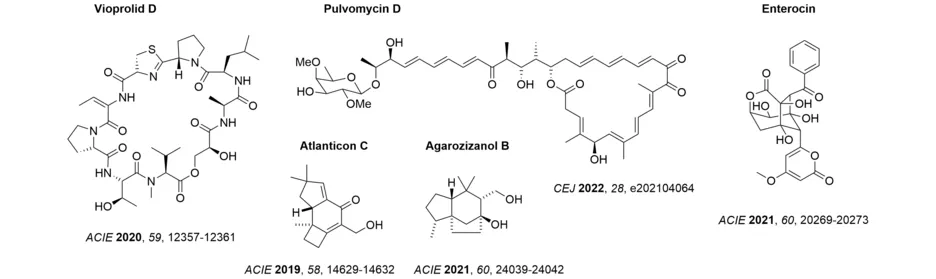
Für unsere Auswahl synthetischer Zielmoleküle gilt die biologische Aktivität einer Verbindung ebenfalls als Kriterium. Unter diesem Aspekt arbeiten wir an der Synthese von cancerostatischen und antiinfektiven Verbindungen. Exemplarisch seien die Synthesen der GE-Faktoren sowie der Amythiamicine genannt, die den bakteriellen Elongationsfaktor EF-Tu hemmen. Viele dieser Studien wurden und werden in enger Zusammenarbeit mit anderen Gruppen ausgeführt. Wir haben beispielsweise zur Aufklärung des Wirkmechanismus einiger synthetischer EF-Tu-Inhibitoren beigetragen (ChemMedChem 2013, 8, 1954-1962) oder wir haben beim Entschlüsseln des Biosynthese-Wegs der Vioprolide mitgeholfen (Angew. Chem. Int. Ed. 2018, 57, 8754-8759). In neuerer Zeit gelang uns die Erstsynthese eines Vioprolids, des Vioprolid D, dessen sterisch anspruchsvolles (E)-Dehydrobutyrinfragment am Ende der Synthese durch eine Isomerisierung der (Z)-Verbindung etabliert wurde. Die Arbeiten zum EF-Tu mündeten in der erfolgreichen Totalsynthese eines weiteren Inhibitors dieses Proteins, des Pulvomycins D, eines polyketiden Naturstoffs. In diesem Fall ermöglichte eine stereoselektive Aldolreaktion den konvergenten Aufbau des Molekülgerüsts (Synthesis 2021, 54, 4246-4262), die Cyclisierung zum Makrolacton erfolgte durch eine Heck-Reaktion. Mit der Synthese des polyketiden Naturstoffs Enterocin wurde an die Arbeiten zum biogenetisch verwandten Wailupemycin B angeknüpft. Durch einen selektiven, biomimetischen Syntheseweg wurde enantiomerenreines Material erhalten, das eine Zuordnung der Absolutkonfiguration erlaubte (Org. Lett. 2022, 24, 6903-6907).
Die Entdeckung einer neuartigen Kaskadenreaktion, bei der bis zu drei photochemische Schritte aneinandergereiht werden, erschloss einen Zugang zu Sesquiterpenen mit einem Protoilludan- oder Prezizaen-Skelett. Die Nützlichkeit der Methode wurde durch Erstsynthesen des Protoilludans Atlanticon C und des Prezizaens Agarozizanol B demonstriert.
Zu unseren Bemühungen im Bereich der Naturstoffsynthese kommen verwandte Forschungsschwerpunkte hinzu, die sich mit der Herstellung neuer Strukturgerüste für die Medizinalchemie (z.B. Angew. Chem. Int. Ed. 2012, 51, 10169-10172; Nat. Commun. 2020, 11, 5621) oder der Entwicklung neuer Sonden für biologische Studien beschäftigen (z.B. J. Am. Chem. Soc. 2018, 140, 2718-2721). Diese Untersuchungen werden meist in Kooperation mit anderen akademischen oder industriellen Einrichtungen durchgeführt.
Katalytische Verfahren
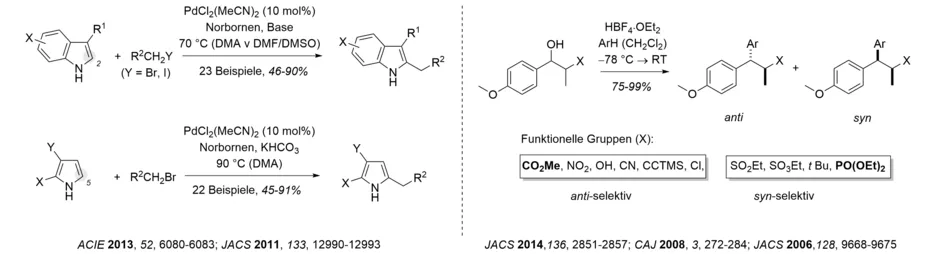
Unser Interesse im Bereich der Heterocyclensynthese führte zur Entwicklung von regioselektiven Kreuzkupplungsreaktionen, die bereits weit verbreitete Anwendungen fanden (Übersicht: Tetrahedron 2005, 61, 2245-2267). In den darauffolgenden Jahren änderte sich der Fokus hin zur einer direkten C-C-Bindungsknüpfung an Heterocyclen durch C-H-Aktivierungsreaktionen. Darunter fallen die Alkylierung von Indolen und Pyrrolen und die Arylierung von Thiophenen (z.B. Chem. Eur. J. 2015, 21, 18407-18416).
Die faciale Diastereoselektivität in intermolekularen Reaktionen von freien Carbeniumionen wurde zum ersten Mal umfassend und systematisch durch unsere Gruppe untersucht. Es konnte gezeigt werden, dass α-chirale benzylische Kationen mit hoher Diastereoselektivität reagieren und dass der Ausgang der Reaktion vom sterischen Anspruch des jeweiligen Substituenten am stereogenen Zentrum in α-Position zum Kation abhängt. Katalytische Varianten dieser Transformationen verwenden FeCl3, AuCl3 oder Bi(OTf)3 als Katalysatoren, außerdem wurde die Methodik auf allylische und propargylische Kationen ausgeweitet (z.B. Angew. Chem. Int. Ed. 2008, 47, 10106-10109).
Nach ersten Arbeiten zur Wasserstoffbrücken-vermittelten Katalyse von enantioselektiven photochemischen Reaktionen (siehe unten) hat sich unser Interesse in diesem Gebiet auf die enantio- und regioselektive Übergangsmetallkatalyse ausgedehnt (Übersicht: J. Org. Chem. 2019, 84, 8815-8836). Template mit Bindestellen zur Substratkoordination wurden entworfen, die gleichzeitig das Anbringen eines katalytisch aktiven Übergangsmetalls wie Mn, Ru oder Rh ermöglichen. Grundlagenuntersuchungen zur selektiven Epoxidierung mit einem Ru-basierten Oxidationskatalysator und Chinolon-basierten Olefinen zeigten zweifelsfrei, dass die Wasserstoffbrückenbindung sowohl für die hohe Regio- als auch für die Enantioselektivität verantwortlich ist. Die Rh-Katalyse ermöglichte eine enantioselektive Durchführung von Aminierungs- und Aziridinierungsreaktionen, Mn-basierte Katalysatoren zum ersten Mal hoch regio- und enantioselektive Oxygenierungsreaktionen (Angew. Chem. Int. Ed. 2018, 57, 2953-2957; Chem. Sci. 2020, 11, 2121-2129).
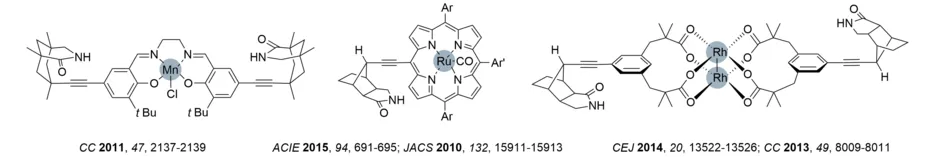
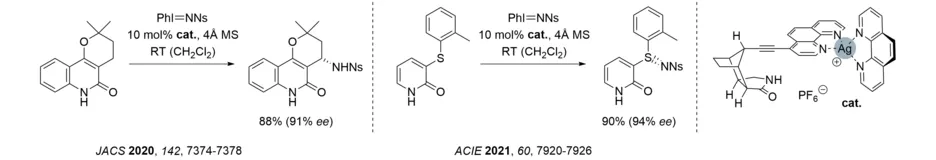
Durch die kovalente Anbindung eines Phenanthrolins an das erprobte Octahydro-1H-4,7-methanoisoindol-1-on-Skelett gelang die in situ Herstellung eines heteroleptischen Silberkomplexes. Dadurch konnten erstmal mit Hilfe der Silberkatalyse enantioselektive Aminierungen und Sulfimidierungen durchgeführt werden (Ns = p-Nitrophenylsulfonyl). Die Substrate waren entweder Chinolone oder 2-Pyridone.
Da ein Hauptziel unserer Forschung die Entwicklung von praktikablen Synthesemethoden ist, versuchen wir stets eine direkte Relevanz nachzuweisen. Beispiele hierzu finden sich in einer einfach anwendbaren C-H Aminierung (Adv. Synth. Catal. 2016, 358, 2083-2087) und einer neuen Pyrrolsynthese (J. Org. Chem. 2016, 81, 6149-6156), die beide in der Synthese heteroaromatischer Alkaloide verwendet wurden, sowie in der Rh-katalysierten Aromatenhydrierung zur diastereoselektiven Herstellung gesättigter 2,5-Diketopiperazine (ACS Catal. 2022, 12, 3628-3633).
Photochemie
Unsere Gruppe beschäftigt sich seit ihrem Bestehen mit photochemischen Fragestellungen. Dabei haben wir uns seit dem Jahr 2000 auf die Herstellung enantiomerenreiner Verbindungen mit Hilfe photochemischer Methoden konzentriert und fundamentale Beiträge zur enantioselektiven Photokatalyse geleistet. Basierend auf dem von der Kemp‘schen Trisäure abgeleiteten 1,5,7-Trimethyl-3-azabicyclo[3.3.1]nonan-2-on-Gerüst konnten wir ein chirales Templat für photochemische und radikalische Reaktionen entwickeln, welches seine vielseitige Anwendung in den letzten 20 Jahren erfolgreich unter Beweis gestellt hat.
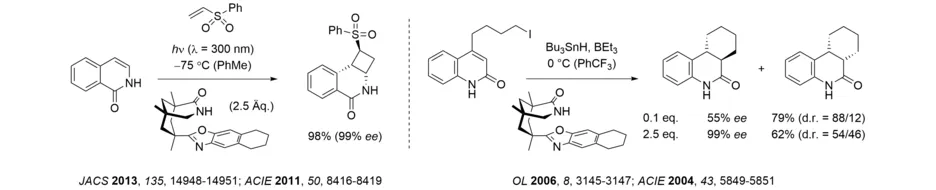
Interessanterweise zeigte das Templat auch katalytische Aktivität in enantioselektiven Radikalreaktionen. Im Bereich der photochemischen Anwendungen sei hier als jüngeres Beispiel der erfolgreiche Einsatz in enantioselektiven intra- und intermolekularen [2+2]-Photocycloadditionen von Isochinolonen genannt.
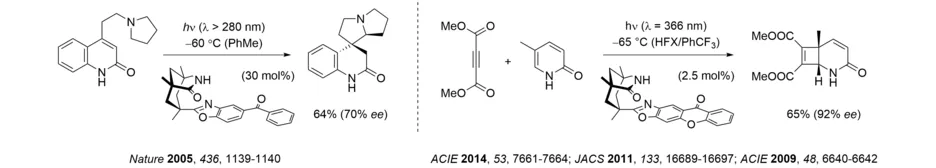
Nach Modifikation des Oxazol-Rückgrats konnte das Templat zu enantioselektiven Katalysatoren für Elektronen- oder Energietransfer (Übersicht: Chem. Rev. 2022, 122, 1626-1653) weiterentwickelt werden. Beispielsweise diente ein Benzophenon als Katalysator in der enantioselektiven Photoredoxcyclisierung eines Aminoethylchinolons. Ein Xanthon konnte für die enantioselektive Sensibilisierung für [2+2]-Photocycloadditionen mit Katalysatorbeladungen von nur 2.5 mol% eingesetzt werden (HFX = 1,3-Hexafluorxylol).
Eine neuere Entwicklung auf diesem Gebiet betrifft die Sensibilisierung durch ein Thioxanthon, die sehr effizient mit künstlichem sichtbarem Licht oder mit Sonnenlicht ausgeführt werden kann. Ein chirales, Wasserstoffbrücken-bildendes Thioxanthon gestattet eine inter- und intramolekulare [2+2]-Photocycloaddition von Chinolonen sowie bemerkenswerterweise die Deracemisierung von Allenen. Die letztgenannte Reaktion vermittelt einen thermisch unmöglichen Prozess, nämlich die Bildung eines einzigen Enantiomers aus einem racemischen Gemisch und hat großes Potenzial für weitere Anwendungen. Die Wirkungsweise beruht auf einem komplexen Zusammenspiel einiger Parameter wie der Assoziation an den Katalysator, der Effektivität der Sensibilisierung und des Zerfalls des jeweiligen Intermediats.
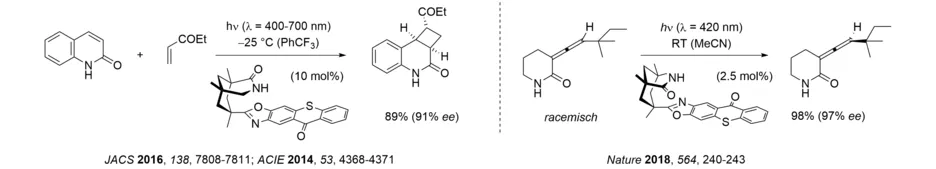
Seitdem wir 2018 zum ersten Mal über eine photochemische Deracemisierung berichtet haben, hat sich das Gebiet sehr schnell entwickelt. In unserer Gruppe liegt der Fokus auf der Entwicklung von Reaktionen, bei denen der Katalysator im ersten, photochemischen Schritt zwischen den beiden Substratenantiomeren unterscheidet. Dadurch dass im Idealfall nur ein Enantiomer prozessiert wird und dabei eine Racemisierung eintritt, reichert sich das andere Enantiomer an, ohne dass Material verloren geht. Bei axial chiralen Allenen und Alkenen kommt es durch Triplettenergietransfer zur Bildung eines achiralen Intermediats, das nach Rückkehr auf die Grundzustandshyperfläche wieder eines der beiden Enantiomere bildet. Wir konnten auf diese Weise sowohl acyclische Allenamide als auch axial chirale Alkene deracemisieren.
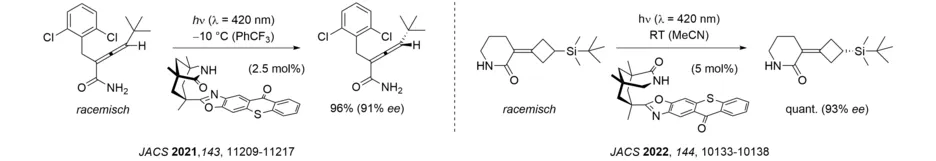
Um eine photochemische Deracemisierung an stereogenen Kohlenstoffzentren vornehmen zu können, müssen reversibel Einfachbindungen gebrochen werden. Wir konnten zeigen, dass Cyclopropane durch Energietransfer eine solche Spaltung eingehen (Angew. Chem. Int. Ed. 2019, 58, 3538-3541), und konnten das für die Deracemisierung von Spirocyclopropyloxindolen verantwortliche 1,3-Diradikal detektieren. Durch reversiblen Wasserstoffatomtransfer zu einem chiralen Benzophenonkatalysator lassen sich Hydantoine mit hohen Ausbeuten und Enantioselektivitäten durch eine photochemische Deracemisierung gewinnen.
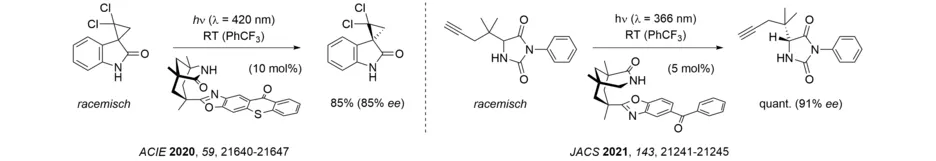
Bereits im Jahr 2010 zeigten wir am Beispiel der intramolekularen [2+2]-Photocycloaddition von Cumarinen, dass sich enantioselektive Photoreaktionen mit Hilfe einer Lewis-Säure-vermittelten Katalyse durchführen lassen. Im Jahr 2013 fanden wir, dass eine derartige Katalyse nicht nur auf Reaktionen von Cumarinen beschränkt ist, sondern überraschenderweise auch auf Enone als Substrate in der [2+2]-Photocycloadditionsreaktion ausgeweitet werden kann. Das Ergebnis ist dahingehend unerwartet, als dass Enone, anders als Cumarine, eine unkatalysierte [2+2]-Photocycloaddition bei einer Wellenlänge von λ = 366 nm eingehen können. Der Mechanismus im letzten Fall unterscheidet sich dabei deutlich von Cumarin-Photocycloadditionen. Die enantioselektive Lewis-Säure-Katalyse von Enon-[2+2]-Photocycloadditionen ist breit anwendbar und wurde bereits neben Dihydropyridonen auch auf 2-Cycloalkenone als Substrate angewandt (Übersicht: Acc. Chem. Res. 2020, 53, 1933-1943).

Es konnte gezeigt werden, dass der Nachteil einer hohen Katalysatorbeladung überwunden werden kann, wenn das Substrat, das nicht an die Lewis-Säure gebunden ist, und der Lewis-Säure-Komplex keine spektrale Überlappung im langwelligen Bereich des UV-Vis-Spektrums aufweisen. Falls diese Bedingung erfüllt ist, bleibt die racemische Hintergrundreaktion aus und die Enantioselektivität bleibt auch bei einer niedrigen Katalysatorbeladung hoch. Die durch sichtbares Licht vermittelte, intermolekulare ortho-Photocycloaddition an Phenanthren-9-carboxaldehyd illustriert dieses Vorgehen. Eine ähnlich niedrige Katalysatorbeladung lässt sich realisieren, wenn die Reaktion auf der Singuletthyperfläche verläuft und eine Hintergrundreaktion des Enons über das entsprechende Triplett ausgeschlossen werden kann. Ein solcher Prozess wurde bei der Oxadi-π-Methanumlagerung von 2,4-Cyclohexadienonen beobachtet und zur enantioselektiven Synthese von Cyclopropanen genutzt.
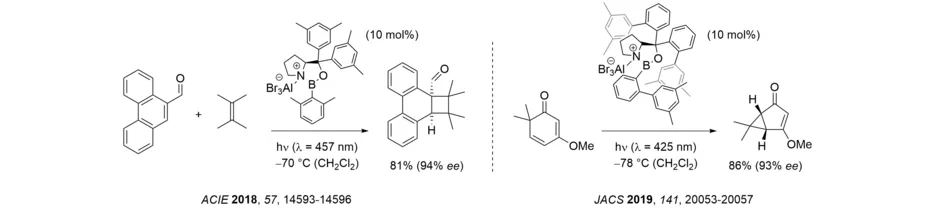
Die letztgenannten Beispiele zeigen, wie ein Chromophor für eine selektive Reaktion bei großer Wellenlänge aktiviert werden kann, und Untersuchungen in diese Richtung werden weiterhin in unseren Laboren durchgeführt (Übersicht: Angew. Chem. Int. Ed. 2018, 57, 14338-14349). Nach der Entdeckung einer Brønsted-Säure-katalysierten [2+2]-Photocycloaddition (Angew. Chem. Int. Ed. 2017, 56, 4337-4341) wurde u.a. gezeigt, dass die zwischenzeitliche Bildung eines Iminiumions einen Triplett-sensibilisierten Reaktionspfad eröffnet, und dass intermolekulare [2+2]-Photocycloadditionen von Iminiumionen enantioselektiv ablaufen. Der Energietransfer vom Rutheniumkomplex (bpy = 2,2‘-Bipyridin) auf das Imininiumion wurde durch zeitaufgelöste Spektroskopie nachgewiesen. Generell wurde gezeigt, dass Iminiumionen eine niedrigere Triplettenergie haben als die ihnen zugrunde liegenden Carbonylverbindungen, wodurch eine Anregung durch Energietransfer möglich ist. Eine chirale Phosphorsäure wurde erfolgreich für die katalytische [2+2]-Photocycloaddition von N,O-Acetalen genutzt, bei der ebenfalls Iminiumionen als Intermediate durchlaufen werden.
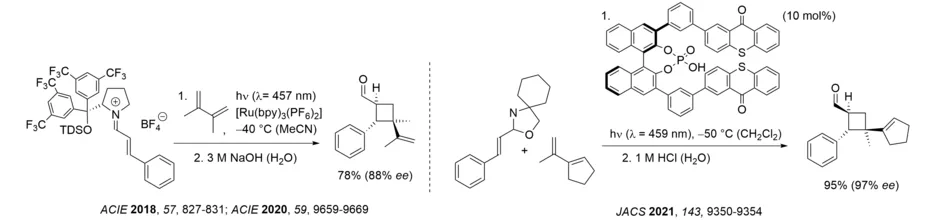
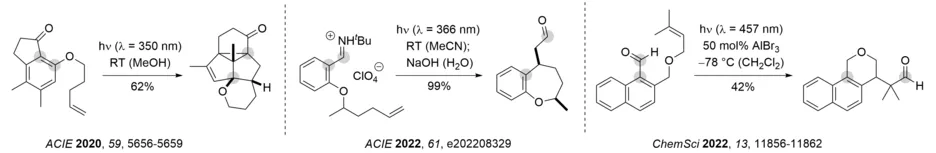
Eine der bemerkenswertesten Eigenschaften photochemischer Reaktionen besteht darin, Molekülgerüste zugänglich zu machen, die thermisch gar nicht oder sehr schwer zu erhalten sind. In diesem Zusammenhang beschäftigen wir uns mit photochemisch induzierten Reaktionskaskaden, bei denen mehrere photochemische Schritte aneinandergereiht werden oder ein photochemischer Schritt eine nachfolgende Umlagerung ermöglicht. Gezeigt sind unten von links nach rechts eine Dreiphotonenkaskade ausgehend von einem 1-Indanon, eine formale intramolekulare Carboformylierung eines Olefins, sowie die Lewis-Säure-katalysierte Bildung von Benzoisochromanen aus 1-Naphthaldehyden. In allen drei Fällen eröffnet eine ortho-Photocycloaddition die jeweilige Reaktionskaskade. Die Reaktion der Iminiumionen kann auch mit sichtbarem Licht und einem geeigneten Sensibilisator durchgeführt werden. Die Reaktionskaskade der 1-Naphthaldehyde gelingt mit chiralen Oxazaborolidinen als Lewis-Säuren mit hoher Enantioselektivität und deutlich höheren Ausbeuten.
Auf dem Gebiet der Photochemie kombinieren wir synthetische Experimente sehr ausgiebig mit photophysikalischen Studien, die teilweise innerhalb unserer Gruppe, teilweise in Zusammenarbeit mit anderen Gruppen durchgeführt werden. Neben den bereits oben genannten Beispielen sind besonders die Arbeiten zur Lewis-Säure-Aktivierung zu erwähnen. Dabei wurden im Zusammenspiel von Synthese, Spektroskopie und Theorie die photochemischen Reaktionen des 2-Cyclohexenon-BF3- (Angew. Chem. Int. Ed. 2021, 60, 10155-10163) sowie des Benzaldehyd-BCl3-Komplexes im Detail aufgeklärt (J. Am. Chem. Soc. 2022, 144, 18927-18937).
Förderung
Unsere Forschung wird durch die TU München und das Land Bayern gefördert. Drittmittel erhalten wir aktuell vorwiegend über den Transregio 325 (https://crc325.de) sowie durch den Gottfried Wilhelm Leibniz-Preis. Beide Programme werden von der Deutschen Forschungsgemeinschaft (DFG) koordiniert und finanziert. Bis Dezember 2020 erhielten wir Unterstützung durch den European Research Council (ERC) im Rahmen des Horizon 2020 Rahmenprogramms (grant agreement No 665951 – ELICOS). Weitere Institutionen, die unsere Forschung über die Jahre fördern oder gefördert haben, sind die DFG (Normalverfahren, GRK 1626), der Fonds der Chemischen Industrie, die Alexander von Humboldt-Stiftung, der Deutsche Akademische Austauschdienst (DAAD), das Elitenetzwerk Bayern, das Bundesministerium für Bildung und Forschung (BMBF) sowie die Studienstiftung des Deutschen Volkes. Mit vielen Unternehmen arbeiten oder arbeiteten wir vertrauensvoll an gemeinsamen Projekten im Rahmen von Forschungskooperationen, unter anderem mit (in alphabetischer Reihenfolge): AstraZeneca, BASF, Bayer, Bicoll, Evonik, Medigene, Novartis, Roche, Sanofi und Wacker.