A-1 - Katalytische anti-Markovnikov Hydratisierung terminaler Alkine
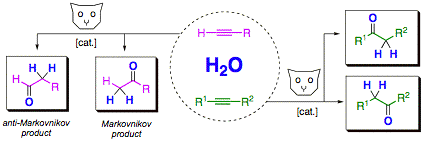
Die katalytische anti-Markovnikov Addition von Wasser an terminale Alkine ergibt synthetisch wertvolle Aldehyde in einem direkten, atomökonomischen Schritt (Review: Synthesis 2007, 1121). Die Reaktion ist deshalb sehr interessant für die organische Synthese. Wir haben in den vergangenen Jahren Katalysatorsysteme für die Reaktion entwickelt, die infolge ihrer hohen Aktivität und einfachen Handhabung erste Anwendungen in der Synthese ermöglicht haben.
Neue Katalysatoren für die anti-Markovnikov Hydratisierung terminaler Alkine
Wir haben einen leicht handhabbaren und hoch aktiven in situ Katalysator für die anti-Markovnikov Hydratisierung terminaler Alkine entwickelt, der sich in der Synthese hervorragend bewährt hat (Org. Lett. 2006, 8, 5853).
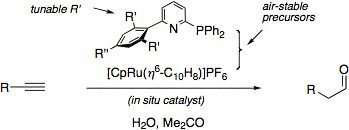
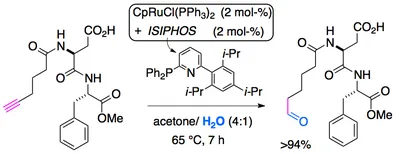
Einen nochmals verbesserten in situ Katalysator für die anti-Markovnikov Hydratisierung terminaler Alkine haben wir aus CpRuCl(PPh3)2 und dem Ligand ISIPHOS in wässrigem Aceton erhalten (J. Am. Chem. Soc. 2011, 133, 8138).
anti-Markovnikov Hydratisierungen in der Organischen Synthese
• Eine iterative Synthese von oligo-1,4-Diolen mit Aldehyd-Endgruppen wurde vorgestellt (Synthesis 2007, 2809).
• Die katalytische anti-Markovnikov-Hydratisierung von N-Tosyl-propargylamiden zu beta-Tosylamido-aldehyden wurde beschrieben (J. Org. Chem. 2007, 72, 5704).
• Unser Katalysator ist durch andere Arbeitsgruppen in Syntheseprojekten eingesetzt worden, siehe z. B.: A. Fürstner et al (Angew. Chem. Int. Ed. 2011, 50, 8739)
• Wir haben eine iterative Synthesemethode für Aldehyde sowohl als auch Alkine entwickelt, die es unter anderem erlaubt, 13C Markierungen mit Leichtigkeit in Alkylketten einzuführen (mit A. Brunner, in Druck 2015/6).
A-2 - Katalytische Hydroalkoxylierung von Alkenen
Additionen von Alkoholen an Alkene sind schwieriger zu katalysieren und weniger untersucht als Additionen an Alkine. Man kann zwei mechanistische Typen der Reaktion unterscheiden: Mit elektronenarmen Alkenen (Michael Akzeptoren) finden basenkatalysierte konjugierte Additionen statt (oxa-Michael Reaktion), wobei die geringe Nucleophilie der Alkohole limitierend ist.
• Unsere Arbeiten zur asymmetrischen katalytischen oxa-Michael Cyclisierung
Der zweite Typus ist die klassische elektrophile Addition von Alkoholen an nicht aktivierte (tendenziell elektronenreiche) Alkene, die säurekatalysiert über Carbeniumionen verläuft. Die Regioselektivität der Reaktion ist durch die Markovnikov-Regel bestimmt, während die Enantioselektivität bisher nicht kontrolliert werden konnte. Fortschritte in der Lewis-Säure-Katalyse von Hydroalkoxylierungen sind erzielt worden, die Reaktionen weisen aber oft alle Merkmale einer Brønsted-Säure-Katalyse auf, für die wir den Begriff "hidden acid catalysis" eingeführt haben (J. Org. Chem. 2011, 76, 9353) und verlaufen nicht enantioselektiv (Übersicht: Top. Organomet. Chem. 2010, 31, 123).

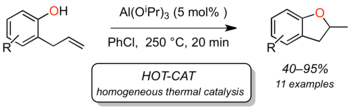
Um den Einfluss von Säurespuren auszuschliessen haben wir als Katalysatoren Verbindungen mit stark basischen Ligand verwendet, und zwar Metall-Alkoxide. Es zeigte sich, dass insbesondere Aluminium(III)isopropoxid bei einer Temperatur von 250 °C den Ringschluss von 2-Allylphenolen zu 2-Methylcumaranen katalysiert (ChemCatChem 2013, 5, 3309):
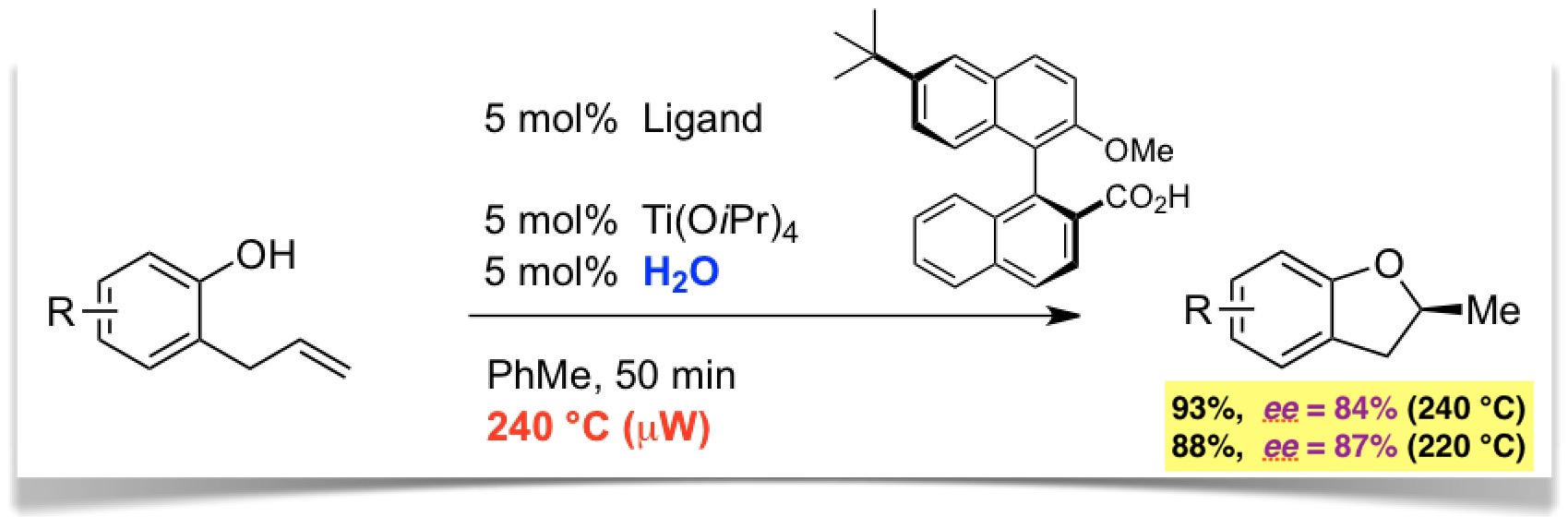
Schliesslich konnten wir zeigen, dass chirale Biarylcarbonsäuren in Kombination mit Titan(IV)isopropoxid und co-katalytischem Wasser (!) die Reaktion asymmetrisch katalysieren (Angew. Chem. 2015, 127, 4086). Die Reaktion läuft bei sehr hoher Temperatur (220-240 °C) mit beträchtlicher Enantioselektivität ab (bis 87% ee) und stellt daher ein Beispiel für "HOT-CAT" (homogene thermische Katalyse) dar.